Vsa živa bitja na Zemlji v svojih celicah skladiščijo informacijo, ki je zakodirana v nukleotidnih zaporedjih nukleinskih kislin DNK in RNK. Ta informacija definira zgradbo/videz, funkcijo in interakcijo celice z ostalimi celicami ali okoljem. Vsako naravno okolje torej nosi informacijo vseh organizmov, ki tam živijo, kar imenujemo okoljska DNK. Zaporedje DNK v organizmih danes je rezultat mutacij in naravnega odbiranja skozi milijone let evolucijske zgodovine prednikov te celice oziroma organizma, ki ga celice sestavljajo. Slovenščina temu v sled pozna zelo ustrezno sopomenko nukleinskim kislinam, t. j. dedni material. To zgodovino lahko s preučevanjem dednega materiala tudi odkrijemo in na ta način ugotovimo, kako so si organizmi med seboj sorodni ali povezani, ali pa preprosto ugotavljamo njihovo identiteto na podlagi referenčnih zaporedij.
Od svojega odkritja so bile raziskave DNK ena temeljnih dejavnosti bioloških laboratorijev. Do komercializacije t. i. metod visokozmogljivostnega sekvenciranja DNK leta 2004 so bile raziskave organizmov na podlagi njihovega dednega materiala vedno omejene s tehnološko učinkovitostjo inštrumentov, ki preberejo zaporedje DNK. To je pomenilo, da je bilo že pridobivanje sekvenc posameznih genov, kaj šele celotnih genomov, zamudno in drago. Za primer: leta 2003 so objavili prvi človeški genom v okviru projekta »Human genome project (Projekt človeškega genoma)«, za katerega so v tistem času porabili približno 3 milijarde ameriških dolarjev in več kot deset let. Današnji sistemi visokozmogljivostnega sekvenciranja, temelječ na tehnologijah, kot so Illumina NovaSeq, Pacific Biosciences, Oxford Nanopore in druge, omogočajo, da lahko človeški genom sekvenciramo že za 1000 ameriških dolarjev in rezultate dobimo takoj. To pa ne bi bilo mogoče, če referenčni človeški genom, pridobljen v sklopu Projekta človeškega genoma, ne bi bil poznan. Referenčni genom je digitalna zbirka podatkov o nukleotidnem zaporedju, ki so jo sestavili znanstveniki kot reprezentativni nabor genov v posameznem organizmu določene vrste. Zato je pridobivanje genomov novih organizmov še vedno relativno mukotrpno in drago početje, medtem ko je primerjava in pridobivanje podatkov za organizme, kjer so reference poznane, lažje. Pod črto: danes lahko preprosto in za relativno nizek vložek pridobimo ogromno genetskih podatkov, ključ do njihovega razumevanja pa je predvsem v poznavanju referenčnih sekvenc, genov, genomov in primerni bioinformatski analizi.
Čeprav se tehnologija visokozmogljivostnega sekvenciranja uporablja v veliki meri za biomedicinske raziskave, so predvsem mikrobiologi in mikrobni ekologi že zelo zgodaj prepoznali njeno uporabnost pri tako imenovanih metagenomskih raziskavah. Tu raziskovalci sekvenciramo okoljsko DNK ali RNK. Lahko gre za celokupno ali pa tarčno pomnoženo DNK, iz katere nato lahko zaradi izrednega števila pridobljenih sekvenc ugotavljamo vrstno sestavo, sestavljamo genome prisotnih mikroorganizmov ali pa se osredotočamo na funkcionalne gene, torej tiste, ki predstavljajo neko biološko funkcijo (npr. dihanje, fotosinteza, nitrifikacija) in iz tega sklepamo, kako mikrobiološko aktivno je neko okolje, od koder je bil vzorec vzet. Na ta način smo v preteklih letih pridobili številne prelomne podatke o mikrobiomih celotnih ekosistemov, pa tudi zelo specifičnih mikrobiomov, kot je na primer mikrobiom človeškega črevesja. Mikrobiom je celokupen nabor mikroorganizmov v določenem okolju. V Alternatorju pa smo že pisali o mikrobiomu slovenskega morja.
Visokozmogljivostno sekvenciranje počasi prodira v uporabo tudi pri višjih organizmih, zaenkrat večinoma še v obliki sekvenciranja pomnoženih tarčnih genov v mešanih ali nepoznanih vzorcih (metabarkodiranje). Pri pomnoževanju tarčnih genov si izberemo določen gen in ga z metodo verižne reakcije s polimerazo (PCR) pomnožimo, da je njegova pojavnost v vzorcu velika in ga tako lažje zaznamo. Največkrat gre za pomnoževanje tako imenovanih filogenetskih označevalcev. To so geni, ki so relativno dobro ohranjeni v vseh organizmih, torej je njihovo nukleotidno zaporedje (sekvenca) precej podobno in nespremenjeno, hkrati pa dovolj različno, da je na podlagi teh zaporedji možno razlikovati med različnimi skupinami organizmov. Ni presenetljivo, da so prav filogenetski označevalci po navadi geni, ki so ključni za preživetje organizma, njihova drastična sprememba pa bi lahko vodila v nefunkcionalnost tega organizma. Tako so najpogosteje uporabljeni filogenetski označevalci tisti geni, ki kodirajo sestavne dele ribosomov (celične strukture, kjer se DNK prepisuje v RNK) ali pa proteine, ki sodelujejo pri celičnem dihanju ali fotosintezi (kot so npr. citokrom c oksidaza ali pa RuBisCo). Ključno pri določanju filogenetske pripadnosti organizmov v mešanih vzorcih je poznavanje referenčnih zaporedij. To so tista zaporedja, ki so bila pridobljena iz znanih osebkov, torej tistih, katerih identiteto smo potrdili z drugimi metodami, recimo z mikroskopijo, in lahko potemtakem predstavljajo referenco, na katero se lahko sklicujemo, ko dobimo neko še neznano, novo zaporedje. Ni odveč povedati, da je ozko grlo vseh metagenomskih ali metagenetskih analiz prav pomanjkanje referenčnih podatkov, saj večina organizmov – še posebej pa to velja za mikroorganizme – ni bila nikoli izolirana in preučena na način, da bi lahko postala referenčna. Mnogih mikroorganizmov namreč sploh ne znamo gojiti. Kljub temu pa se stanje tudi na tem področju bliskovito izboljšuje, tako da vedno bolj(e) spoznavamo ekosisteme in njihove biološke gradnike.
V preteklosti sem v Alternatorju že pisal o pravkar opisanem procesu, torej pridobivanju referenčnih podatkov, in sicer na primeru fitoplanktonskih kremenastih alg v slovenskem morju. V nedavni raziskavi (Turk Dermastia idr. 2023) pa smo šli s sodelavci še korak dlje in znanje o genetski ter morfološki pestrosti lokalne flore kremenastih alg nadgradili z inovativno metodo metabarkodiranja, torej visokozmogljivostnega sekvenciranja določenega pomnoženega gena. Sekvencirali smo gen, ki kodira veliko podenoto encima RuBisCO (rbcL), ključnega encima za asimilacijo ogljika v procesu fotosinteze. Ker imajo vsi fotosintetski organizmi gen za rbcL, so si torej med seboj sorodni in imajo skupnega prednika, ki je prvi posedoval encim RuBisCO. Ker je ta nepogrešljiv za delovanje teh organizmov, se skozi evolucijo ni veliko spreminjal, a vendar dovolj, da se med različnimi vrstami malce razlikuje. Zato velja za dober filogenetski označevalec, vendar do zdaj v raziskavah morskih ekosistemov ni bil uporabljen v nobeni raziskavi, ki bi se posluževala metabarkodiranja. Ker smo v preteklih raziskavah ugotovili, da rbcL zelo dobro razlikuje med posameznimi vrstami kremenastih alg, hkrati pa je dovolj dobro ohranjen, da lahko enostavno zajamemo celoten spekter pestrosti, smo se odločili, da ga uporabimo v omenjeni raziskavi. Potrdilo se je, da je v primerjavi z označevalci, ki se običajno uporabljajo v te namene in temeljijo na ribosomskih genih, rbcL veliko bolj diskriminatoren med vrstami, hkrati pa je zajel ogromno, do zdaj nepoznano pestrost, celo na ravni različnih populacij iste vrste. Z veliko verjetnostjo smo uspeli ugotoviti prisotnost šestih novih rodov in kar dvaindvajset novih vrst za naše okolje, v katerem se fitoplankton kontinuirano spremlja že več kot petdeset let. In to samo na primeru kremenastih alg, medtem ko nas analiza podatkov za ostale fitoplanktonske skupine še čaka. Prednost je tudi že omenjeno razlikovanje na nivoju populacij iste vrste, kar nam ponuja dodaten uvid v ekološko specializacijo in celo evolucijo preprostih enoceličnih organizmov, kot so kremenaste alge. Hkrati se je pokazala že prej omenjena pomanjkljivost, in sicer da številne sekvence niso bile klasificirane do nivoja vrst in celo rodov, saj preprosto vlada pomanjkanje znanja in podatkov v referenčnih zbirkah sekvenc.
S tem se raziskovalci mikroorganizmov soočamo tudi sicer, saj so številne vrste pod mikroskopom, ki je klasično orodje za raziskovanje teh organizmov, preprosto neprepoznavne in jih tudi najbolj izkušeni taksonomi ne morejo klasificirati. Metabarkodiranje je tudi časovno in cenovno učinkovitejše kot klasične metode, je pa res, da je čas od pridobitve vzorca do rezultatov precej daljši. Poleg tega imajo vse analize, ki temeljijo na metabarkodiranju, zaenkrat še veliko pomanjkljivost: popolna kvantifikacija pojavnosti vrst/skupin je nemogoča. Razlog tiči v načinu pridobivanja podatkov, kjer se originalni vzorec pomnoži z metodo PCR, pri čemer pride do eksponentne ojačitve signala bolj pogostih vrst. Tu lahko pride do velikega odstopanja med originalnim vzorcem in pomnoženim vzorcem, pri čemer je kontrola tehnološko zaenkrat neizvedljiva. Deloma lahko ta odstopanja nadomestimo z različnimi normalizacijami podatkov (na primer z logaritemsko normalizacijo), česar smo se poslužili tudi v pričujoči raziskavi, pri čemer smo tudi orali ledino z novejšimi pristopi, vendar kvantifikacija kot taka še vedno ni mogoča. Bolj relevantne podatke glede pojavnosti skupin oziroma vrst lahko pridobimo z metagenomskimi pristopi, kjer ne pride do pomnoževanja tarčnih genov in se sekvencira le originalni mešani vzorec, vendar pa je pri tem treba zagotoviti veliko večjo »globino sekvenciranja«, kar v praksi pomeni, da bomo isti vzorec morali prebrati večkrat in nato podatke združiti, to pa podraži in oteži sam postopek zbiranja podatkov. To je v kontekstu spremljanja vrstne pestrosti in okoljskega monitoringa evkariontskih organizmov zaenkrat še nepraktično. Z metodo metabarkodiranja, ki smo jo uporabili mi, pa je okoljski monitoring že možen in razkrije podrobnosti v biodiverziteti, ki so bile do zdaj neodkrite in neopažene.
-
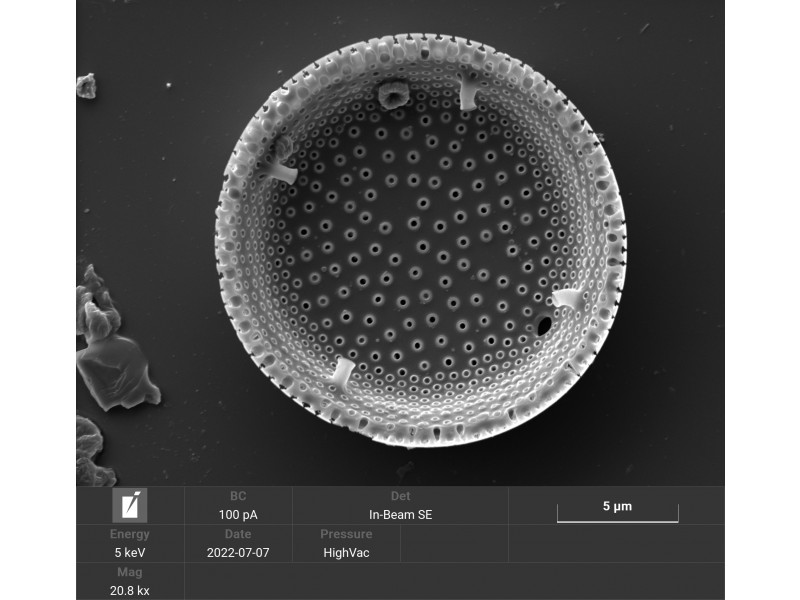 1 / 3
1 / 3 Del silikatne hišice (frustule) kremenaste alge Actinocyclus sp. Slikano z elektronskim vrstičnim mikroskopom (posnetek: Petra Slavinec), pridobljeno v okviru RI-SI-LifeWatch projekta, MIZŠ in ESRR.
-
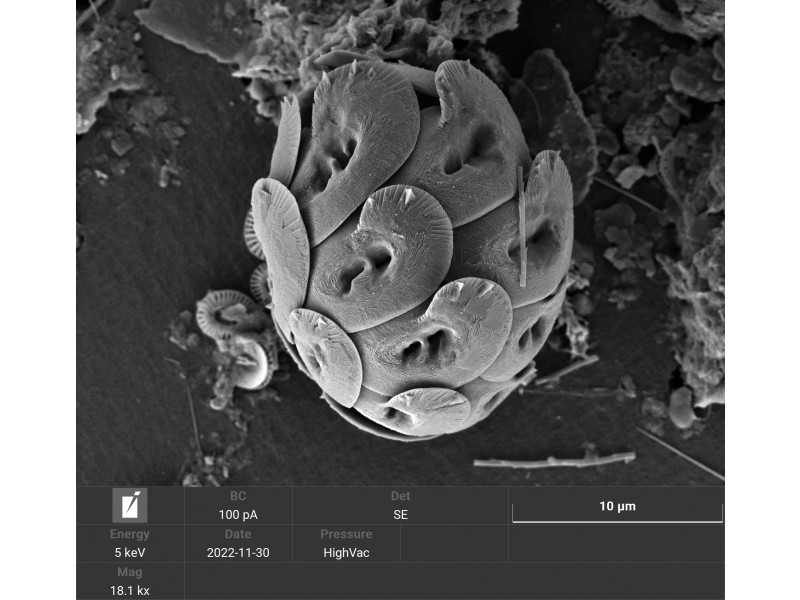 2 / 3
2 / 3 Karbonatno ogrodje kokolitoforida Helicosphaera carteri. Slikano z elektronskim vrstičnim mikroskopom (posnetek: Petra Slavinec), pridobljeno v okviru RI-SI-LifeWatch projekta, MIZŠ in ESRR.
-
 3 / 3
3 / 3 Celulozno ogrodje (teka) ognjene alge (dinoflagelata) Podolampas bipes. Slikano z elektronskim vrstičnim mikroskopom (posnetek: Petra Slavinec), pridobljeno v okviru RI-SI-LifeWatch projekta, MIZŠ in ESRR.